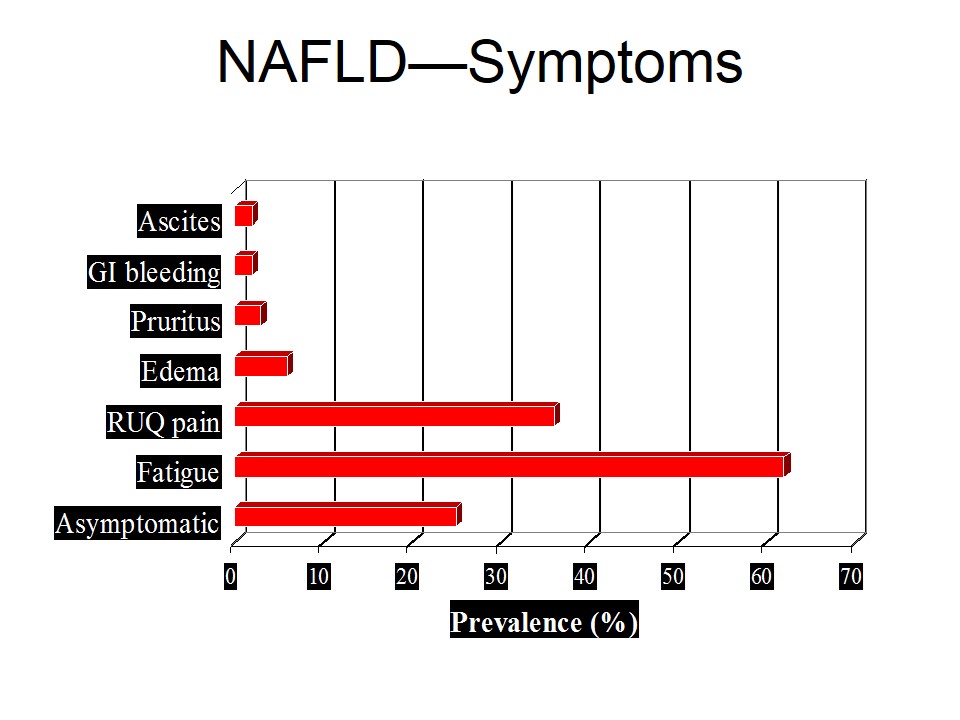
Inborn errors of purine metabolism include the following:
1. Hypoxanthine-guanine phosphoribosyl transferase
Severe deficiency (Lesch-Nyhan syndrome)
The Lesch-Nyhan syndrome is an X-linked recessive disorder, due to severe deficiency of HGPRT, characterized by
● Hyperuricaemia,
● Mental deficiency,
● Spasticity,
● Choreoathetosis
● Self mutilation
The plasma urate level is usually, but not invariably, raised and the urate excretion rate is greatly increased. It is thought that there is acceleration of de novo purine synthesis due to increased availability of 5-PRPP rather than decreased feedback inhibition of PRPP-AT by purine nucleotides.
Partial deficiency
Partial deficiency of HGPRT results in severe X-linked gout but with minor or absent neurological manifestations.
● Gouty arthritis and uric acid lithiasis develop in adolescence or early adult life.
● There is increased urate production as evidenced by hyperuricaemia and increased urate excretion.
2. Adenine phosphoribosyl transferase
Deficiency of APRT occurs as an autosomal recessive disorder associated with the formation of renal calculi composed of this metabolite of adenine.
There is no associated purine overproduction, hyperuricaemia, or gouty manifestations.
3. Phosphoribosylpyrophosphate synthetase
An X-linked disorder resulting in purine overproduction and gout has been described due to excessive activity of PRPP synthetase (PRPPS).
● This increased activity seems to be due to resistance to feedback inhibition by purine nucleotides.
● Gouty arthritis and urate lithiasis associated with hyperuricaemia and increased urinary urate excretion occurs in childhood or early adult life.
4. Adenosine deaminase
Half of the subjects with autosomal recessive severe combined immune deficiency (SCID) have absent ADA activity.
● There is accumulation and increased excretion of adenosine and deoxyadenosine.
● Deoxyadenosine appears to interfere with DNA metabolism.
5. Xanthine oxidase
Deficiency of xanthine oxidase (XO) is an autosomal recessive disorder associated with
● Increased urinary excretion of xanthine and hypoxanthine,
● Xanthine lithiasis,
● Hypouricaemia.
● The only clinical manifestation is the formation of xanthine calculi.
6. Glucose 6-phosphatase
Deficiency of glucose 6-phosphatase (von Gierke’s disease) results in the
● Accumulation of glycogen (liver, kidney),
● Fasting hypoglycaemia,
● Lactic acidosis,
● Hypertriglyceridaemia
● Hyperuricaemia.
● Gouty arthritis usually appears before the age of 10 years and the subjects will go on to develop chronic tophaceous gout if untreated. The hyperuricaemia is due to increased de novo purine synthesis and decreased renal urate excretion (lactic acid inhibition of renal excretion). The increased purine synthesis is probably due to excessive production of ribose 5-phosphate as a consequence of increased shunting of glucose 6-phosphate through the pentose phosphate pathway.